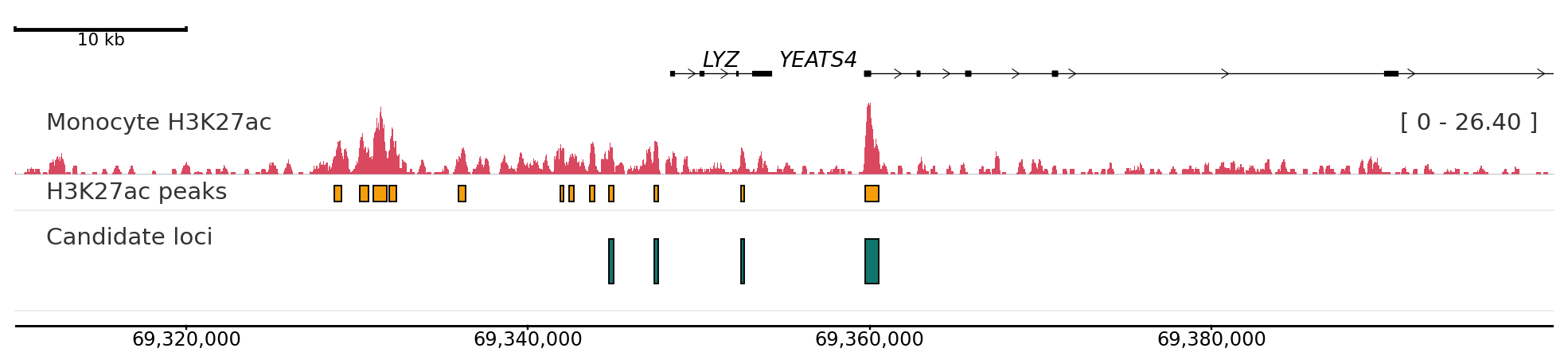
PlotNado builds genome browser-style figures from Python code or editable YAML templates. It is useful when you need reproducible plots for BigWig-like signals, intervals, peaks, links, genes, and optional matrix-style genomic data.
Start with Installation if you are setting up the project, or Quick Start if you already have the uv environment.