Use this when one overlay panel should sit next to ordinary signal tracks without drifting onto a different scale.
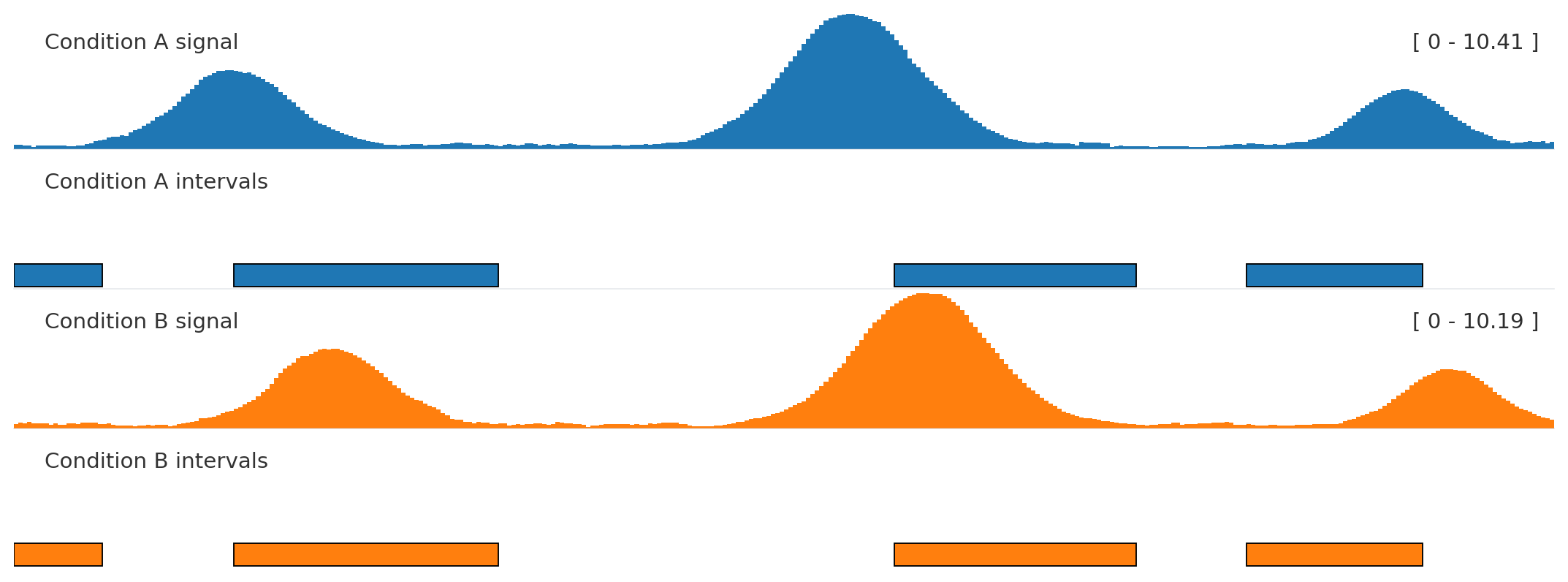
Autocolor + related interval tracks
from plotnado import GenomicFigurefrom plotnado.examples import REGION, intervals, signal# signal() → DataFrame(chrom, start, end, value) — replace with a BigWig path/URL or DataFrame# intervals() → DataFrame(chrom, start, end, name) — replace with a BED/BigBed path, URL, or DataFramefig = GenomicFigure(track_height=1.05).autocolor("tab10")fig.bigwig(signal(phase=0.0), title="Condition A signal", color_group="A")fig.bed(intervals(), title="Condition A intervals", color_group="A", display="expanded")fig.bigwig(signal(phase=1.1), title="Condition B signal", color_group="B")fig.bed(intervals().assign(name=["b1", "b2", "b3", "b4"]), title="Condition B intervals", color_group="B", display="expanded")fig.plot(REGION)
Use the same color_group for tracks that represent the same sample, condition, or assay.
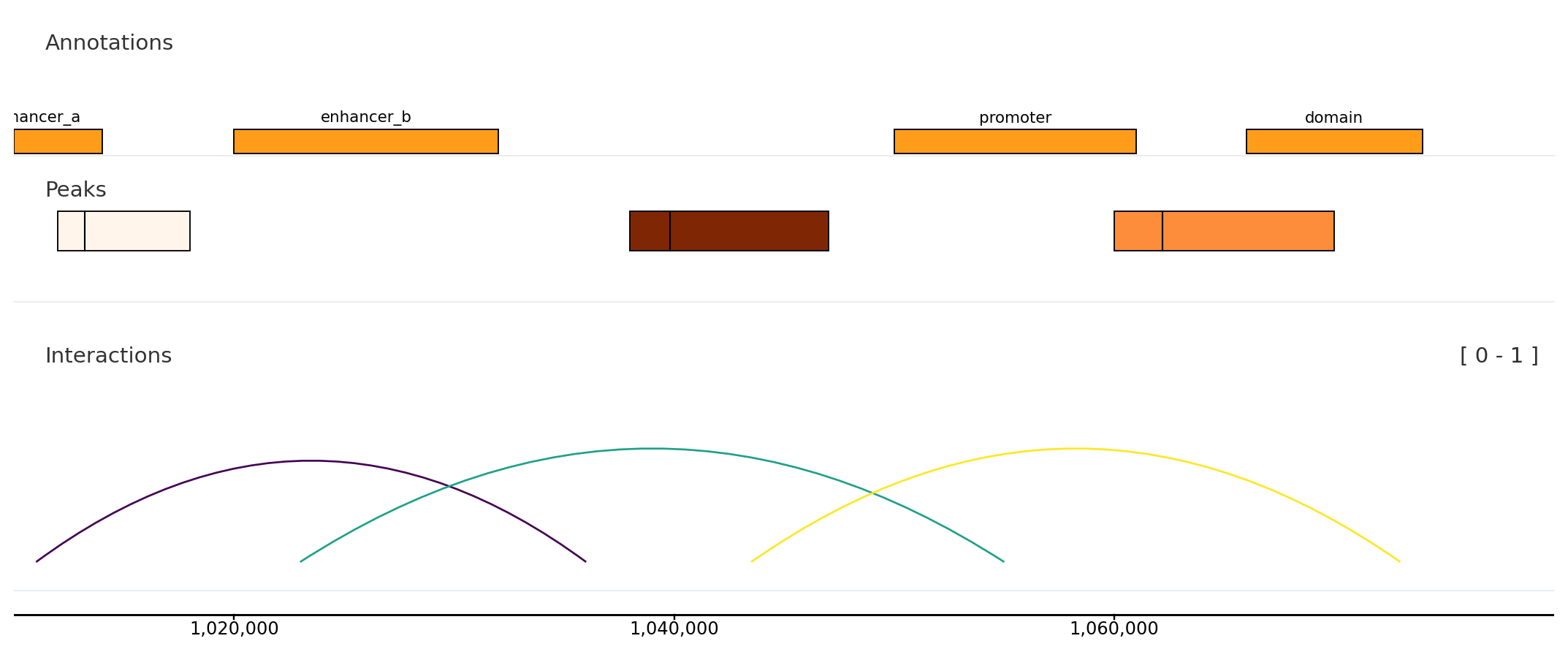
Interval and link context
from plotnado import GenomicFigurefrom plotnado.examples import REGION, intervals, links, narrowpeaks# intervals() → DataFrame(chrom, start, end, name) — replace with a BED/BigBed path, URL, or DataFrame# narrowpeaks() → DataFrame(chrom, start, end, name, score, strand, signalValue, pValue, qValue, peak) — replace with a .narrowPeak path or DataFrame# links() → DataFrame(chrom1, start1, end1, chrom2, start2, end2, score) — replace with a BEDPE path or DataFramefig = GenomicFigure(track_height=1.1)fig.bed(intervals(), title="Annotations", display="expanded", show_labels=True)fig.narrowpeak(narrowpeaks(), title="Peaks", color_by="signalValue", cmap="Oranges")fig.links(links(), title="Interactions", color_by_score=True, cmap="viridis")fig.axis()fig.plot(REGION)
Use interval tracks for local annotations and links() for paired genomic anchors.
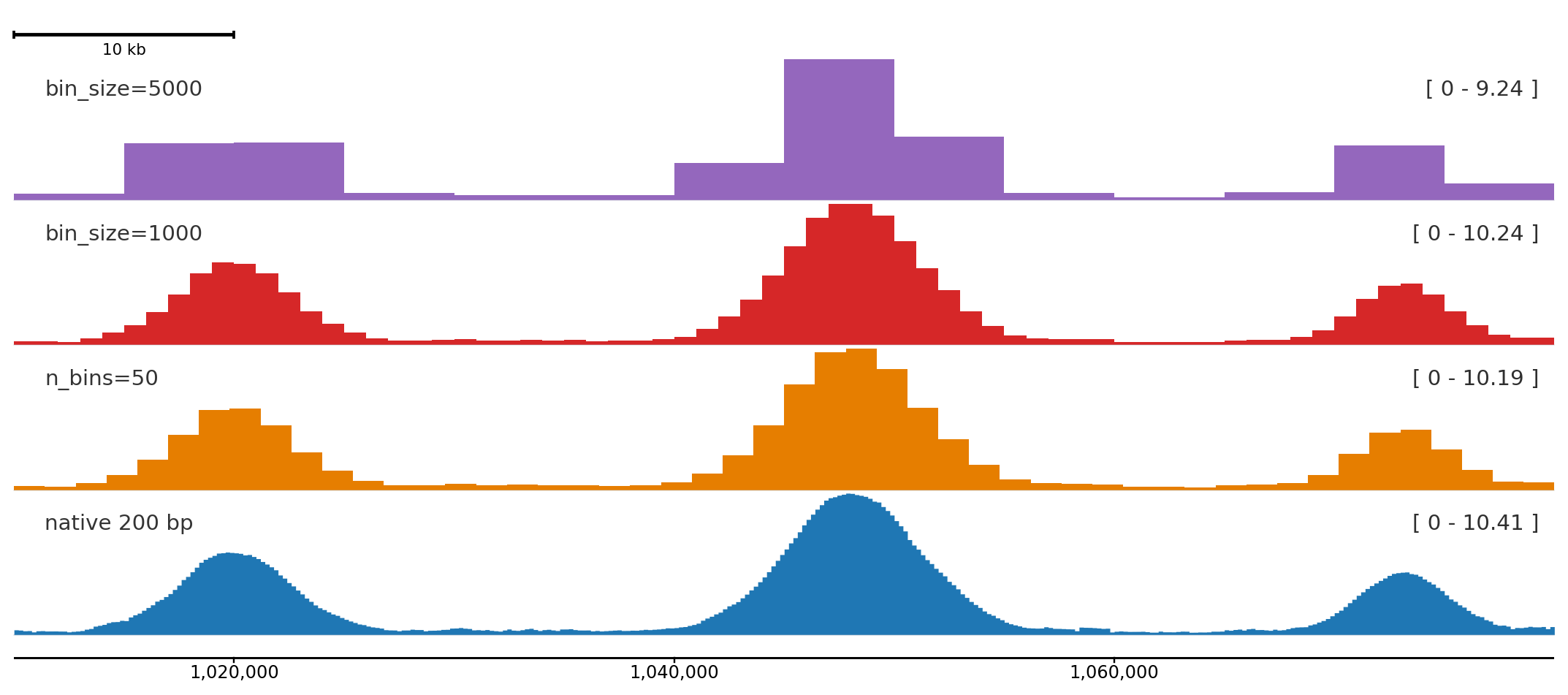
Control signal resolution
Use bin_size or n_bins to match bin width to the figure size and zoom level. Coarser bins reduce visual noise in wide regions; finer bins reveal peak shape at high zoom.
Same signal at three resolutions. bin_size keeps physical bin width fixed in bp; n_bins keeps the count fixed regardless of region size.
For BigWig files the rebinning is applied after fetching the native intervals, so there is no extra file I/O cost. For DataFrames the same weighted-average logic applies.