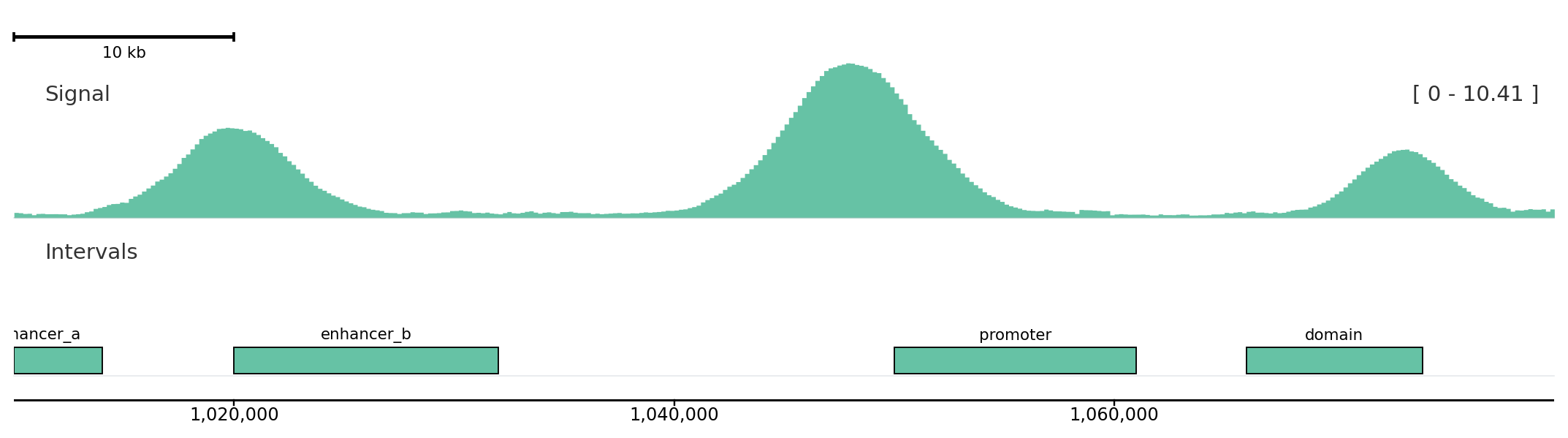
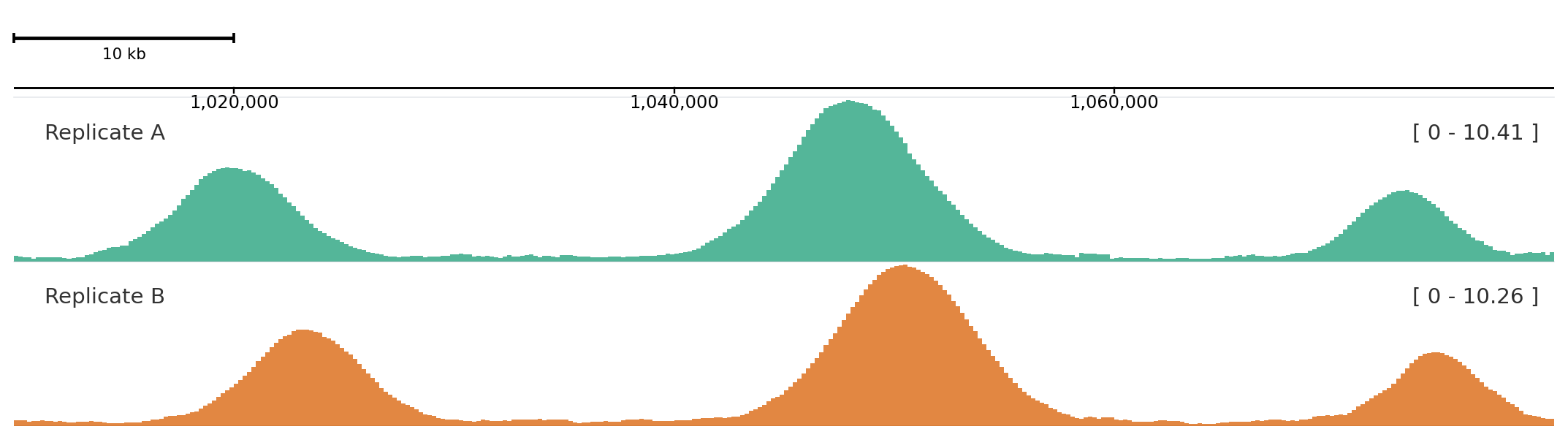
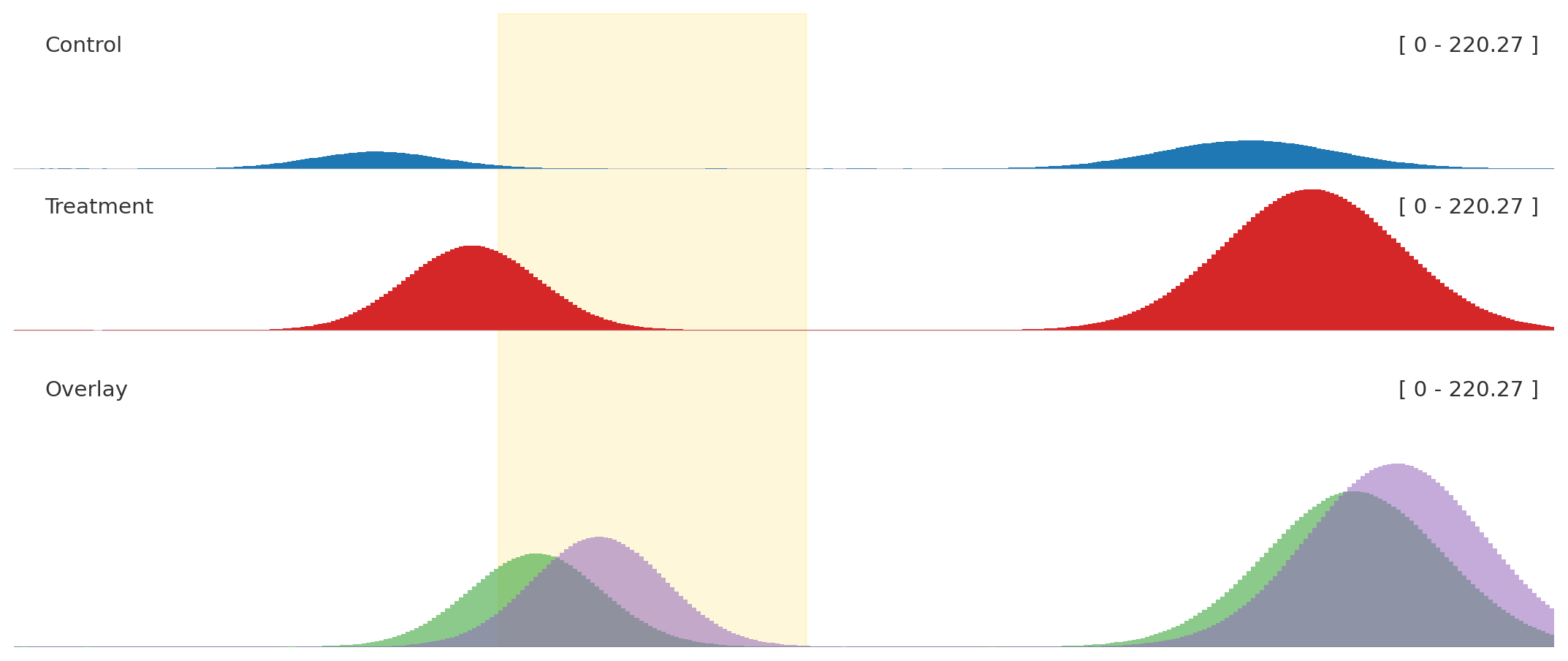
Use helper methods for most figures, add_track() when names come from configuration, and explicit track classes only when you need direct object construction.
Helper methods
from plotnado import GenomicFigurefrom plotnado.examples import REGION, intervals, signal# signal() → DataFrame(chrom, start, end, value) — replace with a BigWig path/URL or DataFrame# intervals() → DataFrame(chrom, start, end, name) — replace with a BED/BigBed path, URL, or DataFramefig = GenomicFigure(track_height=1.2)fig.autocolor("Set2")fig.scalebar()fig.bigwig(signal(), title="Signal", style="fill", color_group="sample-a")fig.bed(intervals(), title="Intervals", display="expanded", show_labels=True, color_group="sample-a")fig.axis()fig.plot(REGION)
Helper methods keep ordinary figure construction readable and chainable.
add_track() is useful when track names are read from YAML, TOML, or another runtime source.
from plotnado import GenomicFigurefrom plotnado.examples import REGION, signal# signal() → DataFrame(chrom, start, end, value) — replace with a BigWig path/URL or DataFramefig = GenomicFigure().autocolor("Dark2")fig.add_track("scalebar")fig.add_track("axis")fig.add_track("bigwig", data=signal(phase=0.0), title="Replicate A", alpha=0.75)fig.add_track("bigwig", data=signal(phase=0.8), title="Replicate B", alpha=0.75)fig.plot(REGION)
Aliases map to the same track constructors used by helper methods.
Explicit objects
Use explicit classes when you need to pass track objects around before adding them.
from plotnado import BigWigTrack, GenomicFigurefrom plotnado.examples import signal# signal() → DataFrame(chrom, start, end, value) — replace with a BigWig path/URL or DataFrametrack = BigWigTrack(data=signal(), title="Reusable signal", style="fill")fig = GenomicFigure().add_track(track)
Ordinary signal tracks and the overlay share one autoscale_group.
Option lookup
from plotnado import GenomicFigureGenomicFigure.available_track_aliases()GenomicFigure.track_options("bigwig")GenomicFigure.track_options("overlay")GenomicFigure.track_options_markdown("genes")